Table of Contents
ELaSTIC is a software suite for a rapid identification and clustering of similar sequences from large-scale biological sequence collections. It is designed to work with data sets consisting of millions of DNA/RNA or amino acid strings, using various alignment criteria. Thanks to the clever use of the sketching technique, and the application of carefully engineered parallel algorithms, ELaSTIC is extremely efficient and scalable while maintaining sensitivity. Thanks to its modular design, it can be easily combined with other tools, like MCL, for the downstream analysis. At the core of ELaSTIC is an efficient MinHash-based strategy to detect similar sequence pairs without aligning all sequences against each other.
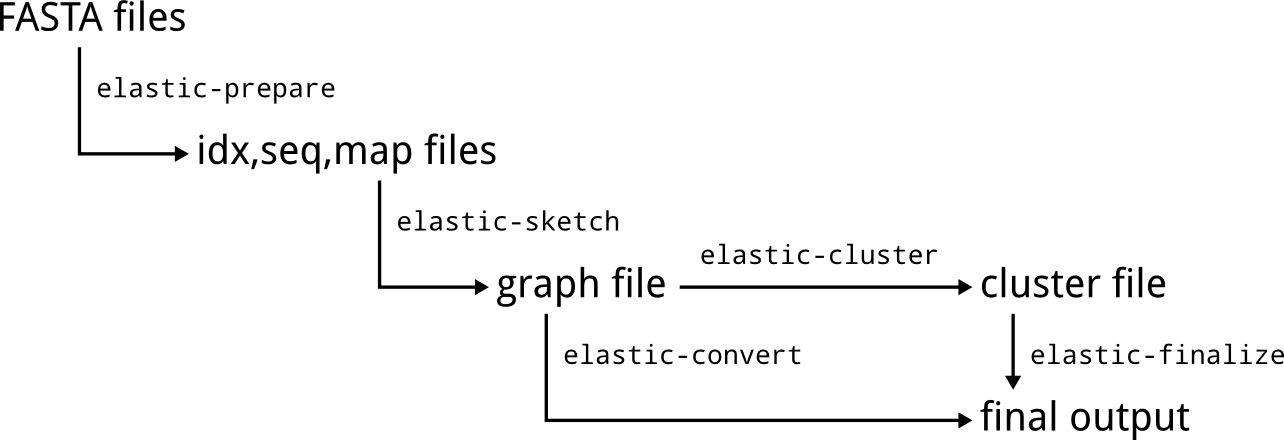
ELaSTIC consists of a set of sequential and parallel tools that can be organized into an end-to-end similarity detection and clustering workflow. Figure 1 provides a general overview of how data flows between different ELaSTIC tools. In the most common scenario, input FASTA files are preprocessed using elastic-prepare to remove redundant and low quality sequences, and to create index files. Usually, this is a one-time effort. The resulting index files are then used as an input to elastic-sketch, which is the main computational workhorse. Typically, elastic-sketch will be executed on a computational cluster or a supercomputer for the maximum performance. The obtained similarity graph can be next converted into a human-readable form with elastic-convert. Alternatively, the analysis can be continued with a clustering tool such as elastic-cluster or mcl, followed by elastic-finalize to obtain a human-readable clusters representation.
ELaSTIC is designed to run on Linux and POSIX systems, which dominate on the current computational clusters. All tools in the ELaSTIC suite are called via an intuitive command line interface, and are optimized for batch processing and inclusion in workflow scripts.
ELaSTIC exploits different forms of parallelism depending on the task. Specifically, elastic-prepare benefits from OpenMP and can run efficiently on multi-core workstations and servers. For example, in our tests we used a server with 16 cores. OpenMP is not required to run elastic-prepare. On the other hand, elastic-sketch is meant for computational clusters and supercomputers and uses MPI. In our tests we ran elastic-sketch on 64 up to 8192 cores of the IBM Blue Gene/P. Note that MPI is necessary to compile and execute elastic-sketch. The remaining tools in the ELaSTIC suite are not computationally demanding and hence run sequentially.
ELaSTIC has been implemented in C++03, OpenMP-2.5 and MPI-2 standards, and uses a subset of the Boost C++ Libraries. Any standard conforming C++ compiler can be used to compile ELaSTIC. We tested GNU C++ 4.6 and newer, Clang/LLVM 3.1 and newer, as well as the IBM XL C/C++ for Blue Gene/P, V9.0. In all cases we found GNU C++ to be generating the most efficient code.
ELaSTIC depends on the Boost C++ Libraries, version 1.48 or newer. To compile all tools except of elastic-sketch, three compiled Boost libraries are necessary: Boost.Filesystem, Boost.IOstreams and Boost.System. elastic-sketch depends on header-only libraries, e.g. Boost.Tuple. Note that all major Linux distributions provide Boost by default. Otherwise, the Boost libraries can be obtained from http://www.boost.org/. Finally, to build elastic-sketch, which usually will be done on a cluster, MPI implementation compatible with the MPI-2 standard and with MPI I/O support is required. Examples include MPICH and Open MPI, and many hardware vendors provide MPI libraries derived from one of these implementations.
To build ELaSTIC, download the latest source code from http://www.jzola.org/elastic
and follow instructions in the doc/INSTALL file:
ELaSTIC - Efficient LArge Scale Taxonomy Independent Clustering
1. Compatibility
This software has been implemented in C++, OpenMP-2.5 and MPI-2 standards.
It has been tested with all major C++ compilers and MPI implementations,
including on platforms such as the IBM Blue Gene. In practice, any standard
conforming C++ compiler and MPI library can be used seamlessly.
2. Requirements
The main requirement are the Boost C++ Libraries, version 1.48 or newer. To
compile all tools except of `elastic-sketch', three compiled Boost libraries
are necessary: Boost.Filesystem, Boost.IOstreams and Boost.System.
`elastic-sketch' depends on header-only libraries, e.g. Boost.Tuple. Note that
all major Linux distributions provide Boost by default. Otherwise, the Boost
libraries can be obtained from http://www.boost.org/. Finally, to build
`elastic-sketch', which usually will be done on a cluster, MPI implementation
compatible with the MPI-2 standard and with MPI I/O support is required.
Examples include MPICH (http://www.mpich.org/) and Open MPI
(http://www.open-mpi.org/). Note that many hardware vendors provide MPI
libraries derived from one of these implementations.
3. Building and installation
The ELaSTIC package consists of five tools: `elastic-prepare', `elastic-sketch',
`elastic-cluster', `elastic-convert' and `elastic-finalize'. It is possible to
build all five tools in one step, or build `elastic-sketch' and the remaining
tools separately. Note that the later option is advantageous if
`elastic-sketch' has to be deployed in a cross-compiled environment, e.g. the
IBM Blue Gene.
3.1. Basic installation
To build all ELaSTIC tools make sure that the Boost C++ Libraries are installed,
and MPI compiler is available in your PATH. If you plan to build only
`elastic-sketch' then MPI and header Boost libraries are sufficient. Otherwise,
compiled Boost libraries must be provided. If you want to build only tools
other than `elastic-sketch' MPI is not required. ELaSTIC uses the CMake build
system: if you are familiar with `cmake' you will find the entire procedure
very easy, if you are not, you will still find it easy. To proceed, unpack
ELaSTIC-X.Y.tar.bz2 tarball, where X and Y are major and minor release numbers.
Enter the resulting directory:
$ tar xfj ELaSTIC-X.Y.tar.bz2
$ cd ELaSTIC-X.Y
Enter the build directory and run `cmake':
$ cd build/
$ cmake ../
This will run basic configuration scripts to detect libraries and compilers.
Once configuration is completed you can build and install all tools by running
`make':
$ make
$ make install
By default, ELaSTIC will be installed in `/usr/local'. You can specify an
alternative installation prefix by passing `-DCMAKE_INSTALL_PREFIX=path'
to `cmake', for example:
$ cmake ../ -DCMAKE_INSTALL_PREFIX=/opt/ELaSTIC
3.2 Building individual packages
By default, `make' will build all five ELaSTIC tools. To disable
`elastic-sketch' you can pass `-DWITH_SKETCH=off' to `cmake':
$ cmake ../ -DWITH_SKETCH=off
Note that this will remove MPI dependency from the configuration script. In a
similar way you can disable all tools other than `elastic-sketch' by passing
`-DWITH_TOOLS=off':
$ cmake ../ -DWITH_TOOLS=off
This will remove compiled Boost libraries dependency as they are not required
by `elastic-sketch'. Keep in mind that combining both options makes no sense.
3.3 Customizing compiler and compiler flags
To specify your preferred C++ compiler you can pass `-DCMAKE_CXX_COMPILER=c++'
to `cmake', for example, to use Clang/LLVM:
$ cmake ../ -DCMAKE_CXX_COMPILER=clang++
In a similar way you can tune compiler flags by passing
`-DCMAKE_CXX_FLAGS=flags'.
3.4 Non-standard Boost installation
If Boost libraries are installed in a non-standard location you will have to
explicitly specify the correct path. You can use `-DBOOST_ROOT=path' to specify
Boost root directory, or `-DBOOST_INCLUDEDIR=path' to specify Boost headers
location and `-DBOOST_LIBRARYDIR=path' to specify compiled libraries location.
3.5 Compiling in a cross-compiled environment
Usually, we find cross-compiled environments on specialized architectures such
as e.g. the IBM Blue Gene. Cross-compilation requires explicitly specifying
compiler, compiler flags and most likely Boost directory. Moreover, in most
cases you will be compiling `elastic-sketch' only, since only this tool is
designed for distributed memory machines. Below is an example of how to build
ELaSTIC on the IBM Blue Gene/P with GNU C++ 4.7 compiler, and Boost header
libraries installed in /opt/boost:
$ cmake ../ -DWITH_TOOLS=off \
-DCMAKE_CXX_COMPILER=powerpc-bgp-linux-c++ \
-DCMAKE_CXX_FLAGS="-static -std=c++11 \
-mcpu=450fp2 -mstrict-align \
-fno-strict-aliasing -O3" \
-DBOOST_ROOT=/opt/boost
3.6 Common problems
The most common problem is incorrectly specified path to Boost libraries.
If you encounter this message:
Use -DBOOST_ROOT=path to specify alternative location
make sure that you have Boost installed, and that you are setting `-DBOOST_PATH'
correctly. Also, keep in mind that if `cmake' options are changed the build
directory must be cleaned before calling `cmake' again:
$ cd build/
$ rm -rf *
4. Copyright
ELaSTIC (c) 2012-2016 Jaroslaw Zola under the MIT License.
BIO (c) 2012-2014 Jaroslaw Zola under the Boost Software License.
JAZ (c) 2004-2014 Jaroslaw Zola under the Boost Software License.
MPIX2 (c) 2005-2014 Jaroslaw Zola under the Boost Software License.
In this section we will demonstrate one example use scenario for ELaSTIC.
We will assume that we have two machines available: server,
a Linux server on which ELaSTIC tools have been installed,
and cluster, a Linux cluster with the SLURM
job manager on which elastic-sketch has been installed. Furthermore,
we will assume that all ELaSTIC tools are included in PATH and can be invoked without specifying their full path.
While this setup may differ from your particular configuration it is general enough to demonstrate
the main ideas.
In our scenario we will generate a similarity graph for a small collection of 16S rRNA sequences from the Human Microbiome Project.
Before we start, let us first download and prepare the test data for this tutorial:
server$ wget ftp://public-ftp.hmpdacc.org/HM16STR/by_sample/SRS066188.fsa.gz server$ gzip -d SRS066188.fsa.gz server$ grep V3-V5 -A1 SRS066188.fsa > SRS066188_V3-V5.fsa
Briefly, we are using wget to download SRS066188.fsa.gz to server,
then we are decompressing the data, and extracting reads derived from the V3-V5 16S rRNA region. ELaSTIC is able
to handle compressed and raw FASTA files, and how you prepare your FASTA files will be specific to your experiments,
and is beyond the scope of this tutorial.
The first step in the ELaSTIC workflow is to make input FASTA files ready for ELaSTIC tools. This is done by running elastic-prepare. The preparation phase has two primary goals. First, to convert raw FASTA files into an indexed binary format optimized for a fast access using parallel I/O, suitable for e.g. large computational clusters. Second, to perform a simple data cleaning and initial clustering to group (nearly) identical sequences. Note that initial grouping is highly recommended as it greatly reduces computational and memory cost in the subsequent steps without compromising sensitivity.
So let us prepare our data. One important factor to consider is the length of input sequences. In general, ELaSTIC is designed to work with longer sequences, and for DNA a minimal recommended length is more than 100nt. For proteins, the recommended length is more than 25aa:
server$ elastic-prepare --input SRS066188_V3-V5.fsa --output SRS066188_V3-V5 \
--type nt --length 100 --clean 0 --group 1After printing some useful information this command will create five new files:
SRS066188_V3-V5.edel SRS066188_V3-V5.eidx SRS066188_V3-V5.emap SRS066188_V3-V5.eplog SRS066188_V3-V5.eseq
Here,
SRS066188_V3-V5.edel lists sequences which have been removed because they were too short,
SRS066188_V3-V5.eidx is an ELaSTIC index file, SRS066188_V3-V5.emap stores result
of the initial grouping, SRS066188_V3-V5.eplog stores the provenance data, and finally SRS066188_V3-V5.eseq
stores compressed input data ready for the next processing step. Please, take a look at SRS066188_V3-V5.eplog
to see what kind of information is reported by elastic-prepare.
You should also check SRS066188_V3-V5.edel to see which sequences have been removed and will not be processed.
The next step in the workflow is to run elastic-sketch.
Because this is the most compute and memory intensive step it should be executed on a computational cluster, or a supercomputer.
In our case we are going to use cluster. First, we have to push to cluster required input files. These are .eidx
and .eseq files, the remaining files are not necessary at this stage. So let us transfer our input:
server$ scp SRS066188_V3-V5.eidx cluster: server$ scp SRS066188_V3-V5.eseq cluster:
Again, details of how you transfer your files will differ depending on your cluster or supercomputer configuration (e.g. you may have to use GridFTP or pull the data to a cluster instead of pushing).
The next and the most crucial step is to prepare configuration file for elastic-sketch. To start, copy the example config file attached with the ELaSTIC source code (or available here) to the same location as your input data. For example:
cluster$ cp /path/to/elastic/share/doc/ELaSTIC-1.70/config.example \ SRS066188_V3-V5.config
Then open it in your favorite editor. Lines starting with # or ; are comments and will be ignored by elastic-sketch.
Remaining lines correspond to command line options supported by elastic-sketch,
for example 'type = nt' corresponds to --type nt, and so on. Now let us walk through all parameters:
'type = nt' — our input data consists of DNA reads hence we set 'type = nt' and we ignore 'sigma' and 'compress' options as they apply to proteins.
'method = 4' — to compare sequences we will use the BLAST identity derived from global alignment, you can check methods description for more details.
'kmer = 16' — we set k-mer length to 16, which seems to be well suited for 16S rRNA. In general, longer k-mers will improve computational efficiency at the cost of potentially decreased sensitivity, shorter k-mers on the other hand may improve sensitivity but can add a significant computational overhead. Moreover, setting too short k-mer length will result in many frequent k-mers (i.e. k-mers shared by many sequences) and this, depending on the 'cmax' configuration, may actually decrease the overall sensitivity.
'gaps = [1,-2,-10,-1]' — since we decided to use the alignment based similarity, we should configure alignment parameters. In our case we have 1 for match, -2 for mismatch, -10 for gap opening and -1 for gap extension.
'level = 60' — we set the similarity level to report only sequence pairs with the BLAST identity above 60%.
'factor = 1' — we set 'factor' to report details of the alignment length, score and the number of identities.
The next part of the configuration file contains parameters that directly influence efficiency of the sketching technique. We recommend that you read Algorithms section and check related publications before tuning these parameters.
'modulo = 20' — this parameter affects the density of sampling of each sequence. If it is low many k-mers will be extracted, which may improve sensitivity of the method at the cost of higher memory and CPU usage. A good starting point is to check the length on the shortest input sequence and set 'modulo' such that more than one k-mer will be extracted from that sequence (i.e. length of the shortest sequence divided by 'modulo' is greater than one). Because in our case the shortest sequence is 100nt, setting 'modulo = 20' is reasonable.
'iterate = 5' — we will run five iterations of the sketching phase. Increasing this number will usually improve sensitivity but will increase runtime proportionally, and may increase memory usage. For very large data sets (with millions of sequences) setting this parameter to 7 or 9 is recommended.
'cmax = 15000' — we will mark each k-mer occurring in more than 15,000 sequences as frequent. Frequent k-mers are not used to extract candidate sequence pairs but contribute to their estimated similarity score. Setting this parameter too high will cause rapid memory explosion, especially if your input is large. Setting it too low will significantly decrease sensitivity. If you have a limited memory and are unable to execute the sketching phase, decrease this parameter and increase 'iterate'.
'jmin = 45' — we will use 45% as the threshold for extracting potentially similar sequence pairs. The candidate pairs are extracted based on the estimated fraction of shared k-mers. For the k-mer based validation method (i.e. 'method = 0') it is sufficient to set 'jmin' to the same value as 'level' or just slightly lower. For the alignment based similarity the choice will vary. If 'level' is high (e.g. above 75%) setting 'jmin' 10-15pp lower is sufficient. For the 'level' range below 30% 'jmin' should be selected carefully by e.g. sampling input sequences and comparing the alignment based score with the k-mer based score.
'steal = 1' — we keep work stealing enabled as this is almost always a good idea.
With config file in place we are now ready to run elastic-sketch — the main graph construction procedure.
elastic-sketch is based on the MPI standard and thus requires mpiexec or a similar MPI launcher. Note that different parallel machines may provide different tools (e.g. ibrun, srun, etc.)
and you should consult the documentation of your particular environment for details. Moreover, depending on the type of job manager running on your cluster details of the job submission and job script preparation will vary.
In our case cluster provides MPI integrated with SLURM, hence we create the following simple script and write it to elastic.slurm file:
#!/bin/sh #SBATCH --time=04:00:00 #SBATCH --nodes=32 #SBATCH --ntasks-per-node=4 srun /path/to/elastic-sketch \ --input SRS066188_V3-V5 \ --output SRS066188_V3-V5 \ --config SRS066188_V3-V5.config
Here, we are requesting 128 cores distributed among 32 nodes. The job will be running for at most 4 hours.
We use --input and --output switches to tell elastic-sketch where to find input and where to write output. Note that these options are obligatory, i.e. they cannot be put in the config file. Also, we do not provide file extensions, since ELaSTIC will read and write automatically all critical files with the same prefix. Finally, we use --config to pass configuration file. We can now submit our job:
cluster$ sbatch elastic.slurm
It is impossible to tell how long the execution will take without details of the underlying hardware architecture. However, one or two tests will give you a pretty good idea as to what to expect. In our case the entire process took 3h20m, with the throughput of 42,450 edges validated per second.
Upon successful completion elastic-sketch will create two files:
SRS066188_V3-V5.eslog SRS066188_V3-V5.sim.00000
The first one stores provenance data and some interesting reports about the execution. The second file represents the actual similarity graph, which is ready for the downstream analysis. It is important to keep in mind that the graph file does not include singleton sequences, that is sequences that ELaSTIC found not similar to any other input sequence.
Graphs created by elastic-sketch are stored as text file, but with sequence identifiers instead of sequence names. These files are suitable for clustering using ELaSTIC tools but also using other methods, for example MCL. In our example, instead of clustering the graph, we will convert it to a human readable format.
This operation can be performed on server once the graph file is downloaded from cluster.
The conversion is done by running elastic-convert with the corresponding map file created during the preprocessing stage:
server$ elastic-convert --input SRS066188_V3-V5.sim --output SRS066188_V3-V5.sim \
--map SRS066188_V3-V5.emap --expand 1
This command will generate a new file, SRS066188_V3-V5.sim.tsv, with sequences represented by their actual names, and including all sequences, i.e. even those that were initially grouped.
The presented above postprocessing step is just one of many possibilities. You can use elastic-cluster to enumerate connected components in the graph, including singletons, or you can run any of your favorite graph analysis tools.
This section is not ready yet. If your combination of data, hardware and ELaSTIC is giving you a headache
please contact Jaroslaw Zola <jaroslaw.zola@hush.com>.
In this section we will use the following convention:
nameimplies a file system path or a path prefix.typeimplies a parameter which is a string or an integer.sizeimplies an integer variable.{0|1}implies a binary variable, where 0 means no/false/disabled, and 1 means yes/true/enabled.
This tool performs the initial preprocessing of FASTA files to filter too short, or low quality sequences, to identify duplicate, or nearly identical sequences, and to generate input files ready for the analysis with elastic-sketch. It employs OpenMP parallelism, and can be executed on any server/workstation with a sufficient main memory.
Supported options:
- --input
name Read input sequences from a file or a directory
name. Input must be in the FASTA format, but can be gzip or bzip2 compressed. Ifnameis a directory, all readable, regular as well as compressed, FASTA files in that directory are used.- --output
name Write output to files with a prefix
name. Five files are created:name.eplogname.eidxname.eseqname.emapname.edel- --type
{nt|aa} Set input sequence type. Currently, only DNA/RNA (
nt) and amino acid (aa) sequences are supported. By defaultntis assumed.- --length
size Filter sequences shorter than
size. Filtered sequences are ignored from any further processing, and are not included in.eidxand.eseqfiles. By default100is assumed.- --clean
{0|1} Filter low quality sequences that contain undetermined residues. For DNA/RNA any character different than the standard A,C,G,T/U is considered undetermined. Similarly, any character different than the standard 20 amino acids is undetermined. If this option is disabled, missing characters are replaced with T in DNA sequences, and A in amino acid sequences. By default
1is assumed.- --group
{0|1} Group duplicate or nearly identical sequences. To group such sequences a very fast super-shingling procedure is used. Each identified group is represented by the longest component sequence, and only these representative sequences are included in
.eidxand.eseqfiles. By default1is assumed.
This is the main tool in the ELaSTIC suite. It constructs a similarity graph by detecting sequence pairs with a similarity score above the input threshold. This tool is parallel, and is meant to run on distributed memory systems with MPI, such as clusters or supercomputers.
Supported options:
- --input
name Read input data from files with a prefix
name. Two files must be provided:name.eidxname.eseq- --output
name Write output to files with a prefix
name. Two files are created:name.eslogname.sim.00000name.tim.00000--factoris enabled) with the resulting similarity graph. Please, refer to File Formats for a detailed description of the ELaSTIC file formats.- --config
name Read input configuration from
name- --type
{nt|aa} Set input sequence type. Currently, only DNA/RNA (
nt) and amino acid (aa) sequences are supported. Note that complete sequence preprocessing, e.g. filtering of low quality sequences, is done in the preprocessing stage by elastic-prepare. By defaultntis assumed.- --sigma
type Enable
typeamino acid (compressed) alphabet. Because the sketching algorithm to identify candidate sequence pairs works with k-mers, usually it is advantageous to employ compressed alphabet at this stage (e.g. Dayhoff6). The alphabet can be specified as a list[group1,group2,...,groupN], wheregrpupIis a single amino acid group. For example,--sigma [AGPST,C,DENQ,FWY,HKR,ILMV]defines the Dayhoff6 alphabet with six groups. Alternatively, two predefined alphabets are provided:A20representing the standard amino acid alphabet, andDayhoff6as defined above. By defaultA20, i.e. no compression, is assumed. This option is used only if--type aais enabled.- --compress {0|1}
Use compressed alphabet during the validation stage. If enabled, all sequences are translated into the selected compressed alphabet before validation. By default
0is assumed. This option is used only if--type aais enabled.- --validate {0|1}
Enable validation stage. To eliminate false positive edges from the candidate sequence pairs pool validation should be perfomed. This is the most time consuming step. By default
1is assumed.- --method type
Specify which method should be used to validate candidate sequence pairs. All methods report some form of sequence identity as an integer value in the range [0,100]. Currently, six different methods are supported:
0k-mer fraction identity, where the fraction of shared k-mers between two sequences over the number of k-mers in the shorter sequence is computed. The k-mer size is set via--kmerswitch. The k-mer fraction identity is a very fast method, and it works particularly well with small alphabets. It can capture sequence containment.1CD-HIT sequence identity computed from the exact global pairwise alignment with cost-free end gaps in the shorter sequence.2CD-HIT sequence identity computed from the exact global pairwise alignment.3BLAST sequence identity computed from the exact global pairwise alignment with cost-free end gaps in the shorter sequence.4BLAST sequence identity computed from the exact global pairwise alignment.5BLAST sequence identity computed from the first highest scoring exact local pairwise alignment.6CD-HIT sequence identity computed from the banded global alignment.7BLAST sequence identity computed from the banded global alignment.
Methods
1to5involve computing pairwise alignment with affine gap penalty, which usually is computationally costly, but may be desired in many cases. There is a significant difference in how CD-HIT and BLAST define identity. The former defines identity as (identities/shorter-sequence-length), while the later as (identities/alignment-length). Here, identities is the number of matches in the alignment, and alignment-length is the total length of the alignment without end gaps. Edgar provides some interesting discussion regarding definitions of pairwise sequence identity. By default0is assumed.- --kmer size
Set k-mer size used by the sketching algorithm to identify candidate sequence pairs, and by the k-mer fraction identity method during the validation stage. The size must be in the range
[3,31]. By default16is assumed.- --gaps type
Specify alignment parameters. Parameters can be provided as
[match,sub,gopen,gext]or[path-to-smatrix,gopen,gext]. Here,match,sub,gopen,gextare match score, mismatch score, gap open score and gap extension score, respectively, whilepath-to-smatrixis a path to a substitution matrix in the NCBI BLAST format. By default[1,-2,-10,-1]is assumed. This option is used only if--method typewithtypedifferent than 0 is enabled.- --level size
Set the sequence identity threshold to decide during validation whether two sequences are similar. The level must be in the range
[0,100]. By default75is assumed.- --factor {0|1}
Include intermediate values required to compute sequence identity in the output files. If enabled, instead of a single similarity value the output file will store three integers representing component values from which similarity can be computed. In some cases, having access to such component values is desired (consider, for example, computing the HSSP score correction). The meaning of the actual values depends on the choice of validation method, as summarized in Table 1.
Table 1.
Method Value 1 Value 2 Value 3 0 Number of shared k-mers Number of k-mers in first sequence Number of k-mers in second sequnce 1-2, 6 Alignment score Length of shorter sequence Number of matches/identities 3-5, 7 Alignment score Alignment length Number of matches/identities
Please, refer to File Formats for a detailed description of the ELaSTIC file formats.- --modulo size
Set mod value used to extract sketches by the sketching algorithm. In general, this value should be set such that the expected number of sketches extracted from any input sequence is more than one. The value must be in the range
[1,100]. By default25is assumed.- --iterate size
Set the number of iterations the sketching algorithm should run to identify candidate sequence pairs. It is recommended to run more than
1iteration. The number of iterations must be less than the mod value set by--modulo. By default7is assumed.- --cmax size
Set the threshold value to detect frequent k-mers. A k-mer is considered frequent if it occurs in more than
sizesequences. Note that frequent k-mers are treated differently by the sketching algorithm. By default10000is assumed.- --jmin size
Set sequence identity threshold to extract candidate sequence pairs. Only pairs with the estimated identity above this threshold are reported, and checked during the validation stage. In general, the threshold should be lower than the similarity level set by
--level, and must be in the range[10,100]. By default50is assumed.- --steal {0|1}
Enable work-stealing in the validation stage. Because computing similarity scores during the validation is inherently imbalanced, work-stealing is used to drastically improve performance. Note however, that work-stealing requires very efficient remote memory access (RMA), and hence on some ancient architectures this option may be better disabled. By default
1is assumed.
This tool converts similarity graph into a tab separated file substituting sequence identifiers with sequence names. It is sequential, and can be executed on any server/workstation with a sufficient memory.
- --input
name Read input graph from a file or a directory
name. The graph must be provided as a list of edges stored in a single or multiple files. Ifnamepoints to a directory, all readable files in that directory are processed. Please, refer to File Formats for a detailed description of the ELaSTIC file formats.- --output
name Write output to file
name. The output is written in a tab separated format, where each row represents one edge. The first and the second column are sequence name, and the remaining columns store sequence identity, or three values as defined in Table 1. Note that if--expandis enabled edges for which the actual similarity was not computed (because they were part of a sequence group) will be reported with the similarity of their representative sequences.- --map
name Use sequence map from a file
name. Sequence maps are created in the preprocessing stage by elastic-prepare. Please, refer to File Formats for a detailed description of the ELaSTIC file formats.- --expand {0|1}
Expand sequence groups in the final output. If this option is disabled only the representative sequence for a given sequence id, which really corresponds to a sequence group, is extracted. By default
0is assumed.
This tool performs clustering of similarity graphs. Currently, it supports only connected components enumeration. It is sequential, and can be executed on any server/workstation with a sufficient memory.
Supported options:
- --input
name Read input graph from a file or a directory
name. The graph must be provided as a list of edges stored in a single or multiple files. Ifnamepoints to a directory, all readable files in that directory are processed. Please, refer to File Formats for a detailed description of the ELaSTIC file formats.- --output
name Write output to files with a prefix
name. Two files are created:name.eclogname.cls.00000- --nodes size
Set the number of nodes in the input graph. If the provided number is higher than the actual number of nodes, the extra nodes are treated as singletons.
- --level size
Set the threshold to filter edges that should not be considered in the clustering process. By default
0is assumed.
This tool performs the final postprocessing to unwind clusters, convert their component sequence identifiers to sequence names, and store them in a human-readable form. It is sequential, and can be executed on any server/workstation with a sufficient memory.
Supported options:
- --input
name Read input clusters from a file
name. Clusters must be provided in the MCL format. Please, refer to File Formats for a detailed description of the ELaSTIC file formats.- --output
name Write output to files with a prefix
name. Two files are created:name.eflogname.clust- --map
name Use sequence map from a file
name. Sequence maps are created in the preprocessing stage by elastic-prepare. Please, refer to File Formats for a detailed description of the ELaSTIC file formats.
All tools in the ELaSTIC suite, except of elastic-sketch, work with text files. elastic-sketch uses a portable binary format for performance.
EIDX Portable binary file
storing index of the input sequences from the accompanying eseq file.
The binary format is used to enable fast parallel I/O on large parallel machines.
ESEQ Portable binary file storing compressed input sequences. The binary format is used to enable fast parallel I/O on large parallel machines.
EMAP Text file with mapping between sequence identifiers and sequence names. The file consists of a list of records, where each record is in the form:
id M seq_name_1 seq_name_2 ... seq_name_M
where, id is an integer sequence identifier, M is the number of duplicate or nearly identical sequences represented by a given identifier, and seq_name_I is the name of I-th sequence represented by a given identifier. seq_name_1 is the name of the longest sequence, that is used as a representative sequence for a given group.
EDEL Simple text file listing names of sequences filtered during the preprocessing stage. A single line stores one sequence name.
SIM.00000 Text file storing similarity graph as an edge list. A single line stores one edge in the form:
source_id [space] target_id [tab] weight
where source_id and target_id are integer sequence identifiers, and source_id < target_id.
TIM.00000 Text file storing extended information about similarity graph. A single line stores one edge in the form:
source_id [space] target_id [tab] Value_1 [space] Value_2 [space] Value_3
where source_id and target_id are integer sequence identifiers, source_id < target_id, and Value_I is defined as in Table 1.
CLS.00000 Text file storing a list of clusters in the MCL format. A single line stores one cluster represented as a list of component node identifiers separated by a white space.
CLUST Text file storing a list of clusters in a FASTA-like, human-readable format. Each cluster is stored in the form:
>Cluster_Name seq_name_1 seq_name_2 ... seq_name_M
where Cluster_Name is a unique cluster name, and seq_name_I is the name of I-th sequence in a given cluster.
Please see Figure 1 in the CLOSET white paper
and Algorithm 1 in the ELaSTIC HiCOMB paper.
Symbols used in these publications map to the elastic-sketch options as follows:
t corresponds to --level,
M to --modulo,
Cmax to --cmax,
and Cmin or tmin to --jmin.